医伴旅服务热线:
医伴旅服务热线:


全部名称
舒尼替尼、索坦、sunitinib、Sutent、Sunitix、舒尼替尼苹果酸盐
适应人群
适用于确诊为胃肠道间质瘤、晚期肾细胞癌、高复发风险肾细胞癌(术后辅助治疗)或进展性胰腺神经内分泌肿瘤的成年患者。[ 详情 ]
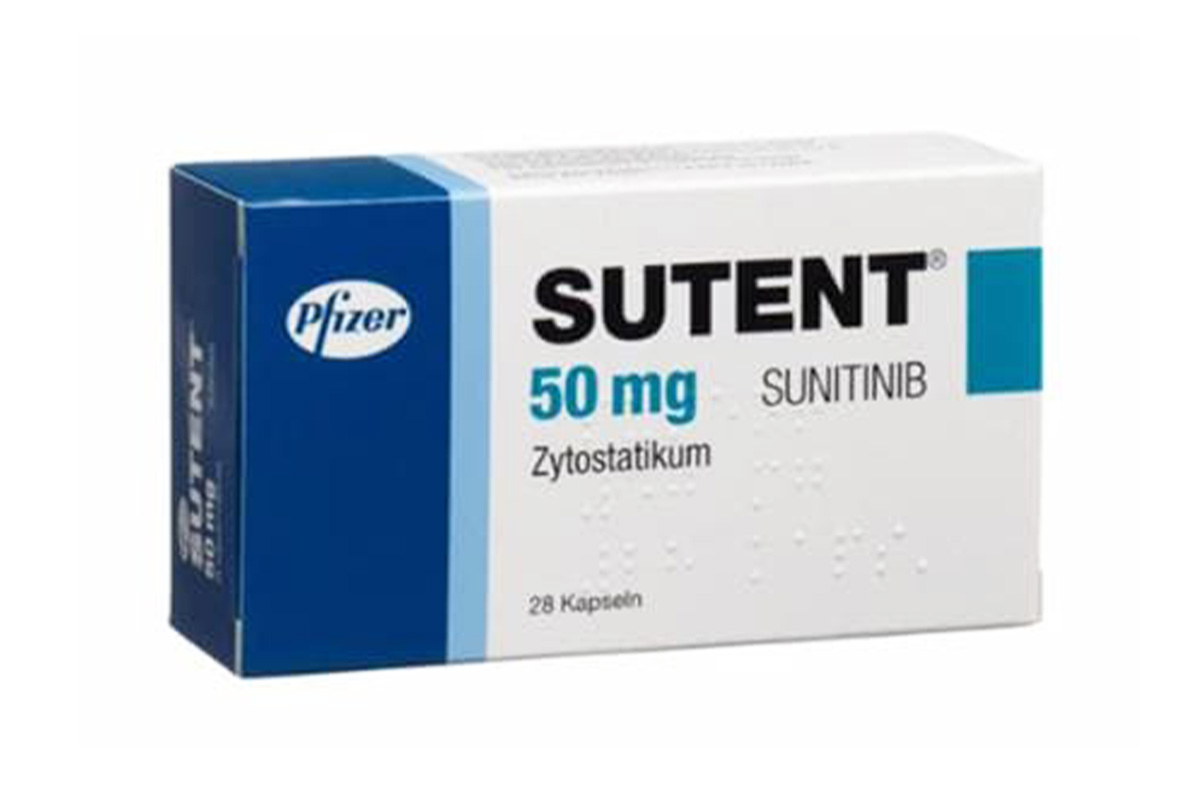


图片来源: 图片来自公开渠道(如FDA官网、Drugs官网、原研药厂官网等),仅供参考。
参考资料: FDA说明书更新于2021年8月,FDA说明书网址:https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=021938

[ 免责声明 ] 本页面内容来自公开渠道(如FDA官网、Drugs官网、原研药厂官网等),仅供持有医疗专业资质的人员用于医学药学研究参考,不构成任何治疗建议或药品推荐。所涉药品可能未在中国大陆获批上市,不适用于中国境内销售和使用。如需治疗,请咨询正规医疗机构。本站不提供药品销售或代购服务。