新药上市是件关乎人类生命健康与安全的大事,必然需要经历一个漫长且严苛的研发审批过程。
第一阶段:科学研发过程
在确认进行新药研制后,由实验室研究寻找关于治疗某种特定疾病的具有潜力的新化合物。这个阶段需持续1-2年时间。
1,寻找药物靶点
任何实验的开展都需围绕药物靶点展开,它是实验的核心点。
2,化合物的筛选与合成
医疗科研学者根据靶点的空间结构,从虚拟化合物库中筛选一系列可匹配的分子结构合成化合物,这被称为先导化合物。
3,活性化合物的验证与优化
在确定先导化合物后,继续进行研究确认。因为不是所有先导化合物都能符合要求,还需通过体外细胞试验验证,初步筛选出活性高、毒性低的化合物,并根据构效关系进行结构优化,这些化合物称为药物候选物。
第二阶段:充分实验并得出结论
实验的主要目的是评估药物的药理和毒理作用,药物的吸收、分布、代谢和排泄情况(ADME)。同时进行生产工艺、质量控制、稳定性等研究(CMC)。这个过程一般需要持续2-4年时间。
实验首先会在动物层面进行展开,确定药物的有效性与安全性,具体研究角度:药效如何?毒副作用如何?是否至畸?是否导致其他突变情况?
在完成动物实验且效果符合预期,会在符合GMP要求的车间完成药物生产。这个过程也非易事,要进行制剂开发,看该药适合如何使用?如果口服吸收效果差就需要开发为注射剂,或遇溶剂性差应如何解决?这些都是制剂过程的关键环节。
第三阶段:人体临床试验
在获得日本临床试验审批后再进行人体临床试验。
人体试验共分三期:
Ⅰ期临床 20-100例,正常人,主要进行安全性评价。
Ⅱ期临床 100-300例,病人,主要进行有效性评价 。
Ⅲ期临床 300-5000例,病人,扩大样本量,进一步评价。
该临床试验过程一般需要3-7年时间。需要科研学者更加详细严谨且全面的记录病人用药后的身体反应及病患恢复情况。且要以安全性为前提。
第四阶段:新药上市审批
在实验成功并符合医药研发要求后即可进行新药上市申报。
申报资料:NDA申报资料 — CTD(Common Technical Document)
CTD主要由五大模块:①行政和法规信息;②概述:药物质量、非临床、临床试验的高度概括;③药品质量详述;④非临床研究报告;⑤临床研究报告
申报流程:
1,批准信
符合要求,可以上市。
2,可批准信
基本满足要求,少数不足可以修改。
申请人应在收到10日内作出回应修正,否则视为自动撤回。
3,拒绝信
存在严重问题或需要补充大量信息资料。
申请人可在10日内提出修正或在30日内要求听证。
NDA特殊审评程序
1,优先审评(Priority Reviews)
适用于能够在治疗、诊断或预防疾病上比已上市药品有显著改进的药品,优先安排NDA审评;
2,加速审批(Accelerated Approval)
用于治疗严重或危及生命疾病的药品,且存在合理并能够测量的“替代终点”(Surrogate endpoint),即药物预期的治疗效果的指标,变通审评标准,利用“替代终点”审评;
3,快速通道(Fast-track)
用于治疗严重或危及生命疾病的药品,且有潜力满足临床尚未满足的医学需求,早期介入,密切交流,分阶段提交申报资料;
审批通过即可上市。
第五阶段:上市后的继续研究
这阶段属于临床监测期,受试者要是IV期临床。数量大于 2000 例,同时要进行社会性考察。待药品上市后4-10年,为保证药品的有效性及安全性还会重新审核 NDA资料。
这就是新药上市的全过程了。看似“小巧”的药剂背后凝聚了数名科研工作者的心血。
2024年日本新药上市明细:
日本药品医疗器械法第十四条中规定的新药定义为:与已经获得上市许可的药品在有效成份、规格、用法用量、适应症等有明确差异的药品,主要包括含新有效成份的药品、新复方制剂、新给药途径、新适应症、新剂型、新用量等。药品若含有已在日本上市的活性成份的酯或盐,也属于含新有效成份。
据日本药品与医疗器械管理局(Pharmaceuticals and Medical Devices Agency,PMDA)新药批准信息,2024年1-2月日本厚生劳动省共批准16个新药上市许可,包括8个化学药品、8个生物制品(含1个疫苗)。4个新药是由日本厚生劳动省全球首次批准上市许可,见表1。
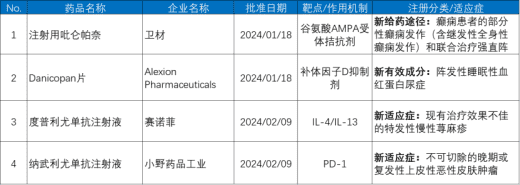
1.注射用吡仑帕奈
2024年1月18日,吡仑帕奈(Perampanel)的新给药途径注射用吡仑帕奈获日本厚生劳动省批准上市许可,用于暂时不适宜口服给药的癫痫患者的部分性癫痫发作(含继发性全身性癫痫发作)和联合治疗强直阵挛性癫痫发作(其他抗癫痫药物疗效不佳时)。吡仑帕奈是卫材公司自主研发的谷氨酸AMPA受体非竞争性拮抗剂。除注射剂外,全球已上市的吡仑帕奈制剂还有片剂口服混悬液。片剂和口服混悬液分别在2019年和2023年获中国NMPA批准上市许可,用于成人和4岁及以上儿童癫痫部分性发作患者(伴有或不伴有继发全面性发作)的治疗。
CAS号:380917-97-5
2.Danicopan片
2024年1月18日,Alexion Pharmaceuticals公司的Danicopan片全球首次获日本厚生劳动省批准上市许可,用于联合其他的补体C5抑制剂治疗阵发性睡眠性血红蛋白尿症。Danicopan(代码:ACH-4471)原研公司Achillion Pharmaceuticals,是一种补体因子D抑制剂,与其他的补体C5抑制剂(Ravulizumab、依库珠单抗)联用时能够通过抑制旁路途径补体级联通路的发生,阻止补体C5激活并对患者自身健康细胞的免疫攻击。Danicopan已获得美国FDA突破性疗法、罕见病用药以及欧盟EMA优先药物(PRIME)计划、罕见病用药的认定。据公司新闻,该药品正在进行单药治疗伴有地图样萎缩的老年黄斑变性的全球多中心Ⅱ期临床试验。
CAS号:1903768-17-1
3.度普利尤单抗注射液
2024年2月9日,赛诺菲公司的度普利尤单抗新适应症获日本厚生劳动省批准,用于现有治疗效果不佳的特发性慢性荨麻疹。该适应症的获批基于1项全球多中心Ⅲ期临床试验(ClinicalTrials登记号:NCT04180488)的结果。度普利尤单抗由再生元和赛诺菲共同研发,是一种拮抗白介素4(IL-4)和白介素13(IL-13)的人源单克隆抗体,2017年美国FDA全球首次批准上市许可,2020年获中国NMPA批准上市许可,中国NMPA批准的度普利尤单抗适应症包括特应性皮炎、结节性痒疹和哮喘。2023年3月,赛诺菲向美国FDA递交度普利尤单抗治疗特发性慢性荨麻疹的上市许可申请。
4.纳武利尤单抗注射液
2024年2月9日,小野药品工业公司的纳武利尤单抗新适应症获日本厚生劳动省批准,用于不可切除的晚期或复发性上皮性恶性皮肤肿瘤。该适应症的获批基于1项研究者主导的日本国内Ⅱ期临床试验结果(日本临床试验登记平台jRCT登记号:jRCT2031190048),纳武利尤单抗由Medarax公司和小野药品工业公司共同研发,是全球首个获批上市许可的PD-1免疫检查点抑制剂,2014年7月在日本全球首次获批上市许可,2014年12月获美国FDA、2015年6月获欧盟EC批准上市许可。2018年6月获中国NMPA批准上市许可(商品名:欧狄沃/OPDIVO),国内已获批治疗非小细胞肺癌、头颈部鳞状细胞癌、胃癌、食管癌、恶性胸膜间皮瘤、尿路上皮癌等9项适应症,2023年5月,纳武利尤单抗治疗上皮性恶性皮肤肿瘤的适应症获得日本厚生劳动省罕见病用药认定。
2023年日本新药上市明细
据日本药品与医疗器械管理局(Pharmaceuticals and Medical Devices Agency,PMDA)新药批准信息,2023年日本厚生劳动省共批准105个新药上市许可,包括52个化学药品、53个生物制品(含13个疫苗),比2022年减少35%。日本厚生劳动省全球首次批准上市许可的22个新药(见表1)中,含新有效成分6个,改良型新药17个,需要说明的是:2023年,第一三共公司的新型冠状病毒mRNA疫苗(Daichirona)既是作为新有效成分、又是作为新适应症获得日本厚生劳动省全球首批上市许可。
2023年关于癌症的新药上市
1.伊布替尼胶囊
杨森公司的伊布替尼(Ibrutinib)胶囊2023年2月24日获日本PMDA全球首次批准新适应症:既往未经过治疗的套细胞淋巴瘤。伊布替尼是布鲁顿酪氨酸激酶(BTK)抑制剂,原研Pharmacyclics公司,2013年获美国FDA全球首次批准上市许可,2014年获欧盟委员会(EC)批准上市许可,2018年获中国NMPA批准进口,中国已获批适应症:用于1)既往至少接受过一种治疗的套细胞淋巴瘤患者的治疗;2)慢性淋巴细胞白血病/小淋巴细胞瘤保留患者的治疗;3)既往至少接受过一种治疗的华氏巨球蛋白血症患者的治疗,或者不适合接受化学免疫治疗的华氏巨球蛋白血症患者的一线治疗;4)联用利妥昔单抗,用于华氏巨球蛋白血症患者的治疗。本次新适应症在日本的获批是基于一项全球多中心Ⅲ期临床试验(登记号:NCT01776840)的结果,2022年3月,杨森公司向欧洲药品管理局(EMA)递交该适应症的上市许可申请,正在审评审批中。
CAS号:936563-96-1
2.锝[99mTc]植酸盐注射液
PDRadiopharma公司的锝[99mTc]植酸盐注射液2023年3月27日获日本PMDA全球首次批准新适应症:用于宫颈癌、子宫内膜癌、头颈癌和外阴癌的前哨淋巴结识别和淋巴闪烁显像。锝[99mTc]植酸盐注射液原研机构富山化学,1977年获日本PMDA全球首次批准上市许可,作为诊断用药用于肝、脾闪烁显像。国内2家企业(广东希埃医药、上海欣科医药)获批该产品上市许可,作为诊断用药主要用于肝、脾及骨髓显像。
3.注射用帕妥珠单抗+曲妥珠单抗+透明质酸酶α
2023年9月25日,日本PMDA全球首次批准了中外制药公司的注射用帕妥珠单抗+曲妥珠单抗+透明质酸酶α新适应症上市许可,用于癌症化疗后进展的HER2阳性、不可切除的晚期或复发性结直肠癌。帕妥珠单抗和曲妥珠单抗均靶向HER2,该复方制剂(代码:RO7198574、商品名:PHESGO)原研为罗氏公司,2020年治疗HER2阳性乳腺癌适应症获美国FDA全球首次批准上市许可,后于同年获EC批准上市许可。日本PMDA批准结直肠癌适应症是基于1项日本国内由研究者发起的开放性单臂Ⅱ期临床研究(日本临床试验登记平台jRCT登记号:jRCT1030220064)的结果。罗氏公司正在中国进行2项该药治疗HER2阳性乳腺癌的Ⅲ期临床研究(登记号:CTR20191171、CTR20232070)。
4.托珠单抗注射液
中外制药公司的托珠单抗注射液的新适应症2023年9月25日获日本PMDA全球首次批准上市许可,用于抗恶性肿瘤治疗时伴随的细胞因子释放综合征(CRS)。该适应症的获批基于2项国际多中心临床研究和1项日本国内临床研究的结果,3项临床研究共入组59例接受抗肿瘤治疗时出现CRS的受试者。托珠单抗由大阪大学和中外制药公司共同研发,是一种白介素6(IL-6)受体抑制剂,2005年日本PMDA全球首次批准该药上市许可,2013年获中国NMPA批准进口,国内获批用于:1)类风湿关节炎;2)全身型幼年特发性关节炎;3)由嵌合抗原受体(CAR)T细胞引起的重度或危及生命的CRS。
5.纳武利尤单抗注射液
小野药品工业的纳武利尤单抗注射液新适应症2023年11月24日获日本PMDA全球首次批准上市许可,用于恶性间皮瘤(恶性腹膜间皮瘤除外)。该适应症的获批基于1项日本国内的Ⅱ期IIT临床试验,共纳入20名受试者。纳武利尤单抗(商品名:OPDIVO)是小野药品工业和Medarex公司共同研发的人源PD-1抗体,2014年获日本PMDA全球首次批准上市许可,后在美国、欧洲和中国上市。2023年2月,纳武利尤单抗治疗恶性间皮瘤的适应症获得日本PMDA的优先审评审批和罕见病药品认定。
6.曲美替尼片
2023年11月24日,诺华公司的曲美替尼片新适应症获日本PMDA全球首次批准上市许可:与甲磺酸达拉非尼胶囊联用,治疗存在BRAF基因突变的复发或难治性毛细胞白血病。BRAF V600基因突变导致BRAF激酶结构性激活,进而导致下游MEK激活,刺激肿瘤生长。曲美替尼可以调节丝裂原激活的细胞外信号调节激酶(MEK)信号,达拉非尼能够抑制BRAF激酶,两药品联合使用对肿瘤抑制作用更佳。美国FDA已批准两药品联合治疗的适应症包括存在BRAF V600E以及V600K基因突变的黑色素瘤、非小细胞肺癌、甲状腺未分化癌、实体瘤和低级别(low-grade)胶质瘤。2019年,中国NMPA批准甲磺酸达拉非尼胶囊和曲美替尼片的上市许可,两药品获批用于联用治疗BRAF V600突变阳性的不可切除或转移性黑色素瘤患者,以及Ⅲ期黑色素瘤患者完全切除后的辅助治疗。
7.碘[123I]苄胍注射液
2023年12月22日,PDRadiopharma公司的碘[123I]苄胍(Iobenguane 123I)注射液新适应症获日本厚生劳动省全球首次批准上市许可,用于诊断帕金森病和路易体痴呆的心脏123I-MIBG闪烁显像。碘[123I]苄胍是一种去甲肾上腺素的放射性类似物,原研公司Fuji film,1992年在日本全球首次获批上市许可,已在美国、欧洲和日本获批用于辅助检测肾上腺和心脏交感神经、神经母细胞瘤、嗜铬细胞瘤的gamma闪烁显像。该药品纳入仿制药参比制剂目录(第六十三批),国内原子高科公司正在进行诊断嗜铬细胞瘤/副神经节瘤适应症的1项Ⅰ期(登记号:CTR20200019)和1项Ⅲ期临床试验(登记号:CTR20180839)。
CAS号:76924-93-1
新药上市是造福患者最直接有效的方式,更多新品也在持续上市中,敬请期待!
祝所有人平安健康。